Entwicklung biometrischer Methoden zur Untersuchung der genetischen Architektur von quantitativen Merkmalen in Kulturpflanzen
Forschungsbericht (importiert) 2010 - MPI für Pflanzenzüchtungsforschung
Die Genetik natürlicher Variation
Trotz der Tatsache, dass die großen Fortschritte, die in der Genetik in den letzten 110 Jahren erzielt wurden, meistens Gene betraf, die für qualitative Merkmale kodieren, wird erwartet, dass der größte Teil der natürlich auftretenden Variation bei Menschen, Tieren und auch in Pflanzen durch geringe genetische Änderungen in einer großen Zahl von Genen, den so genannten quantitative trait loci (QTL), verursacht wird. Der Begriff QTL-Kartierung fasst alle diejenigen Untersuchungen zusammen, die darauf abzielen, einzelne QTL zu identifizieren, deren Einfluss auf das Merkmal zu quantifizieren und ihre Position im Genom zu bestimmen. Das Grundproblem der Untersuchung quantitativer Merkmale ist die Tatsache, dass der Phänotyp eines bestimmten Individuums nur sehr wenig über seinen Genotyp aussagt. Zum Beispiel können zwei Maispflanzen zwei Meter groß, aber trotzdem sehr unterschiedlichen Genotyps sein.
Das Prinzip der QTL-Kartierung
Ein wichtiger Schritt in Richtung QTL-Kartierung war die Entdeckung verschiedener molekularer Markertypen. Mit diesen kann seit den 1980er-Jahren zu immer niedrigeren Kosten der genetische Zustand an einer immer größeren Zahl von Genorten im Genom festgestellt werden. Molekulare Marker können in spaltenden Populationen kartiert werden, das heißt, ihre Position im Genom wird genau erfasst. Wenn nun der Merkmalswert der Individuen der spaltenden Population für ein zu untersuchendes Merkmal erhoben wird, dann können diejenigen Genombereiche identifiziert werden, die zur Variation des untersuchten Merkmals beitragen. Im Gegensatz zu qualitativen Merkmalen reicht das Auge allerdings nicht mehr aus, sondern es müssen statistische Verfahren eingesetzt werden (Abb. 1).


Assoziationsgenetik: Neues Verfahren zur QTL-Kartierung
Der Vorteil des zuvor beschriebenen Verfahrens der kopplungsbasierten QTL-Kartierung ist, dass die Wahrscheinlichkeit, ein vorhandenes QTL zu identifizieren, im Falle der Nutzung einer der genetischen Komplexität des Merkmals angemessenen Populationsgröße sehr hoch ist. Allerdings weist dieses Verfahren auch mehrere Nachteile auf. Einer ist, dass lediglich zwei alternative genetische Zustände (Allele) an jedem Genort im Genom gleichzeitig miteinander verglichen werden können. Es wird allerdings erwartet, dass die Anzahl von Allelen, die in einer Art pro Locus vorkommen, sehr viel größer als zwei ist. Ein weiterer Nachteil der kopplungsbasierten QTL-Kartierung ist, dass die Kartierungsauflösung sehr niedrig ist, das heißt die Genombereiche, in denen das QTL identifiziert wird, sind sehr groß und beinhalten normalerweise mehrere Hunderte bis Tausende von Genen.
Ein neueres Verfahren zur QTL-Kartierung in Pflanzen, das aus der Humangenetik übernommen wurde, ist die Assoziationskartierung. Diese Methode nutzt keine spaltenden, sondern natürliche Populationen oder Kollektionen von Individuen. Die Assoziationskartierung erlaubt eine sehr viel höhere Auflösung sowie den Vergleich einer sehr viel höheren Anzahl von Allelen (Tab. 1).

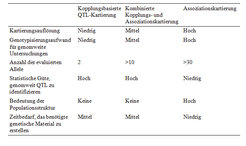
Ursprünglich wurden für die Assoziationskartierung sehr einfache statistische Modelle genutzt, die vergleichbar zu denjenigen der kopplungsbasierten QTL-Kartierung waren [1]. Die genetische Zusammensetzung von Assoziationskartierungs-Populationen ist allerdings sehr viel komplexer, das heißt, es können Subgruppierungen auftreten. Darüber hinaus können sich die Individuen einer Assoziationskartierungs-Population im Hinblick auf ihre familiäre Verwandtschaft unterscheiden [2]. Wenn diese Aspekte bei der statistischen Analyse vernachlässigt werden, wird der beobachtete Prozentsatz an falsch-positiven Assoziationen die nominale Typ-I-Fehlerrate erheblich überschreiten, was wiederum die Interpretation der identifizierten QTL schwierig macht [3]. Daher wurden verschiedenste biometrische Verfahren zur Assoziationskartierung in Pflanzen entwickelt [4, 5], wobei Verfahren, die auf gemischten Modellen beruhen [6], in nahezu allen Fällen die nominale Typ-I Fehlerrate einhalten.
Es gibt zwei Situationen, in denen die Wahrscheinlichkeit, ein QTL mittels Assoziationskartierung zu identifizieren, sehr niedrig ist. Die erste ist, wenn das interessierende Merkmal sehr stark mit der Populationsstruktur korreliert ist. Dies ist der Fall für alle Merkmale unter lokaler Adaptation oder diversifizierender Selektion in verschiedenen Subpopulationen [7]. Die zweite Situation ist, wenn die Allelfrequenzen an den QTL sehr niedrig oder sehr hoch sind. Falls diese Probleme auftreten, kann eine Kombination aus Kopplungs- und Assoziationskartierung für eine QTL-Kartierung erfolgreich sein.
Kombinierte Kopplungs- und Assoziationskartierung
Diese Methode nutzt eine größere Anzahl von spaltenden Populationen, die auf systematische Weise von einer großen Zahl elterlicher Genotypen abgeleitet wurden. Solche Kartierungsansätze, die diese verbundenen Populationen nutzen, versprechen, die Vorteile der kopplungsbasierten QTL-Kartierung mit denjenigen der Assoziationskartierung zu kombinieren [Tab. 1]. Aus diesem Grund haben verschiedene Arbeitsgruppen der Mais-Community gemeinsam eine solche Population erstellt. Diese so genannte nested association mapping Population besteht aus 25 spaltenden Populationen, wobei jede einzelne Population aus 200 Individuen besteht. Der erfolgreiche Einsatz dieser Population, um beispielsweise die genetische Architektur des Blühzeitpunktes bei Mais zu verstehen, wurde kürzlich beschrieben [8].
Die Erstellung derartiger Populationen ist sehr zeit- und kostenintensiv. Daher ist es von außerordentlicher Bedeutung, dass die verfügbaren Ressourcen optimal eingesetzt werden. Dies kann mittels Computersimulationen untersucht werden. Die Ergebnisse solcher Untersuchungen ergaben, dass es vorteilhaft ist, eine möglichst große Anzahl von elterlichen Komponenten zur Erstellung der spaltenden Populationen einzusetzen [9]. Darüber hinaus wurde deutlich, dass selbst in Zeiten, in denen die Erhebung der genotypischen Information mit sehr niedrigen Kosten verbunden ist, die Bedeutung einer präzisen und intensiven Phänotypisierung nicht vernachlässigt werden darf [10].